Bioavailability and bioequivalence studies are essential in the regulatory submission of a new drug product or a generic drug product. Innovator drug product vs Generic drug product. Studies on bioavailability and bioequivalence are necessary to verify therapeutic equivalency. Frequently bioavailability and bioequivalence rely on pharmacokinetics measures such as Area under the Curve (AUC) to assess the extent of systemic exposure and Cmax and Tmax to assess the rate of systemic absorption. You may read: Generic name of drugs, Brand name of Drugs, Generic drug/ medicine, Brand name drug/medicine
Table of Contents
Definition of Bioavailability vs Bioequivalence
According to USFDA, Bioavailability is the rate and extent to which the active ingredient or active moiety is absorbed from a drug product and becomes available at the site of action [1].
According to EMA, The rate and extent to which a substance or its active moiety is delivered from a pharmaceutical form and becomes available in the general circulation (taking into consideration that the substance in the general circulation is in exchange with the substance at the site of action).
Also, according to WHO, The rate and extent to which the active moiety is absorbed from a pharmaceutical dosage form and becomes available at the site(s) of action. Dose, Dosage, Dosage Form, and Dosage Regimen.
Whereas, according to USFDA, Bioequivalence means the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study [1].
A short expression of Bioavailability vs Bioequivalence
BA or F is a short expression of bioavailability. BE is a short expression of bioequivalence.
Which application type requires a BA or BE?
Bioavailability (BA) information of a drug product is essential for Investigational New Drug Application (IND), New Drug Application (NDA), and NDA supplements [2].
On the other hand, a bioequivalence study with pharmacokinetic endpoints of a drug product is essential for Abbreviated New Drug Application (ANDA [3].
The Main focus of Bioavailability vs Bioequivalence
Studies on bioavailability concentrate on figuring out how and when a drug substance escapes from the oral dosage form and reaches the site of action.
On the other hand, bioequivalence studies focus on the performance of the drug product and usually involve comparisons of two drug products: T and R. Here T is the test product. In the United States, R is termed the RLD. In the WHO document, R is termed the “comparator pharmaceutical product” (CPP) [5].
Reference product for Bioavailability vs Bioequivalence
Generally, for the bioavailability study of a new drug product, the reference product should be a solution, suspension, or intravenous (IV) dosage form of the new drug. Conducting a BA study with an intravenous (IV) reference product enables assessment of the impact of the route of administration on BA and defines the absolute BA of the drug released from the drug product. Conducting a BA study comparing one formulation to another enables an assessment of relative BA.
Whereas, for the bioequivalence study of a generic drug product, the reference product should be the innovator drug product or reference listed drug. BE study is to measure and compare formulation performance between two or more drug products. However, in the case of bioequivalence studies of a new formulation of an innovator drug product, the reference product should receive approval. For regulatory approval of a generic drug product (test product, T) must be bioequivalent to the Reference listed drug product R.
What are the objectives of Bioavailability vs Bioequivalence?
Objectives of Bioavailability [2, 6]
- Bioavailability data provide an estimation of the fraction of the drug absorption, distribution, metabolism, excretion, and the effects of food on the absorption of the drug, dose proportionality, or linearity in the pharmacokinetics of the active moieties and when appropriate inactive moieties.
- BA provides useful information to establish dosage regimens and to support drug labeling, such as distribution and elimination characteristics of the drug.
- Bioavailability of drug products aids in the FDA’s evaluation of the safety and effectiveness of a product in IND, NDA, or NDA supplements.
Objectives of Bioequivalence [3, 6]
- To measure and compare formulation performance between two or more drug products.
- Bioequivalence provides a link between pivotal and early clinical trial formulation.
- To determine the therapeutic equivalence between the pharmaceutical equivalence generic drug product and a corresponding reference listed drug.
- For an innovator drug product, bioequivalence studies establish the performance of the new formulation product intended for marketing with an already approved innovator drug product to evaluate the safety/efficacy of the new formulation drug product. Efficacy vs Potency.
- To provide information on product quality and performance when there are changes in components, composition, and method of manufacture after approval of the drug product.
Measurement of Bioavailability vs Bioequivalence
Measurement of Bioavailability
Bioavailability is an indirect or surrogate measure of the rate and extent to which the drug substance or active moiety is absorbed from a drug product and becomes available at its target sites of action. Bioavailability is measured by using three main pharmacokinetic variables:
- The area under the blood drug concentration versus time curve (AUC)
- The maximum blood concentration (Cmax)
- The time to reach maximum concentration (Tmax)
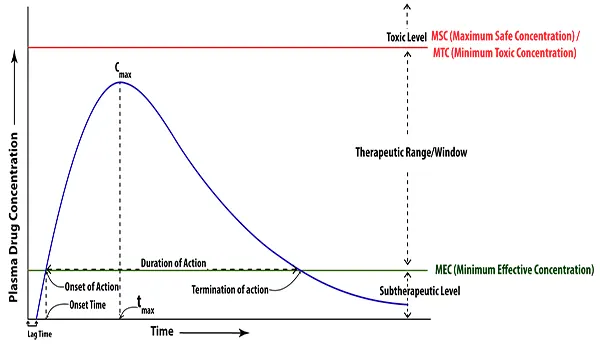
Measurement of Bioequivalence
On the other hand, Bioequivalence (BE) is a surrogate measure of in vivo drug product performance, and dissolution profile comparisons as a measure of in vitro drug product performance. BE has been established via bioavailability testing. Bioequivalence is measured based on the relative bioavailability of the innovator drug product versus the generic drug product.
It is important to note that bioequivalence studies are also performed for innovator drug products in some situations such as: between early and late clinical trial formulations; between formulations used in clinical trials and stability studies, if different; or between clinical trial formulations and to-be-marketed drug products, if different; or between equivalence of product strengths; between marketed innovator product and a new formulation of marketed innovator product (change in one or more excipients).
Bioequivalence is based on a comparison of ratios where the ratio of generic to innovator for each pharmacokinetic variable does not differ by more than 8:10, this is how the range for the confidence intervals is defined: 8/10 = 0.80 gives the lower limit and 10/8 = 1.25 gives the upper limit. The 90% confidence intervals for the ratios of both Cmax and AUC should be contained within the limits of 0.80–1.25 [4]. The FDA considers two products bioequivalent if the 90% confidence intervals (CI) of the relative mean Cmax, AUC (0–t), and AUC (0–∞) of the test (e.g. generic drug product) to RLD (e.g. innovator drug product) should be within 80% to 125% in the fasting state. [1].
Nature of Study
Bioavailability studies are expletory. Whereas, bioequivalence studies are a confirmatory or statutory term that requires ANDA applicants to demonstrate, among other things, that the proposed generic product is bioequivalent to its reference listed drug.
The interrelation between bioavailability and bioequivalence
Bioavailability is not been assessed via bioequivalence testing. Bioavailability is assessed using three main pharmacokinetic variables
- The area under the blood drug concentration versus time curve (AUC)
- The maximum blood concentration (Cmax)
- The time to reach maximum concentration (Tmax)
On the other hand, bioequivalence is assessed via bioavailability testing.
Example of Bioavailability vs bioequivalence
Examples of Bioavailability: after oral administration of 20 mg of Rabeprazole Sodium Delayed-Release tablet, 52% of this drug is absorbed unchanged systematically as compared to intravenous administration. So, the absolute bioavailability of this drug product is 52% [7].
Examples of bioequivalence: two medicines are bioequivalent if there is no clinically significant difference in their bioavailability. Based on the bioequivalence studies under fasting and fed conditions, Idiazole, Rabeprazolnatrium, and Rodesa 20 mg tablets are bioequivalent with Pariet 20 mg. Here, Idiazole, Rabeprazolnatrium, and Rodesa 20 mg tablets are the generic drug product. Pariet is the RLD of Rabeprazole Sodium Delay Release tablet in the UK and Australian markets and Aciphex is the RLD of Rabeprazole Sodium Delay Release tablet for the US market.
Summary of the difference between bioavailability and bioequivalence
| Features | Bioavailability | Bioequivalence |
| Definition | Bioavailability is the rate and extent to which the active ingredient or active moiety is absorbed from a drug product and becomes available at the site of action. | Bioequivalence means the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study. |
| Short expression | BA or F | BE |
| Require for | Investigational New Drug Application (IND), New Drug Application (NDA), and NDA supplements. | Abbreviated New Drug Application (ANDA |
| Main focus | On determining the process and time frame by which a drug substance is released from the oral dosage form and moves to the site of action. | On the performance of the drug product and usually involve comparisons of two drug products: T (test product) and (Innovator drug product or RLD). |
| Reference product | A solution, suspension, or intravenous (IV) dosage form of the new drug. | Innovator drug product or reference listed drug (RLD). |
| Objectives | A. Bioavailability data provide an estimation of the fraction of the drug absorption, distribution, metabolism, excretion, and the effects of food on the absorption of the drug, dose proportionality, or linearity in the pharmacokinetics of the active moieties and when appropriate inactive moieties. B. BA provides useful information to establish dosage regimens and to support drug labeling, such as distribution and elimination characteristics of the drug. C. Bioavailability of drug products aids in the FDA’s evaluation of the safety and effectiveness of a product in IND, NDA, or NDA supplements. | A. To measure and compare formulation performance between two or more drug products. B. Bioequivalence provides a link between the pivotal and early clinical trial formulation. C. To determine the therapeutic equivalence between the pharmaceutical equivalence generic drug product and a corresponding reference listed drug. D. For an innovator drug product, bioequivalence studies establish the performance of the new formulation product intended for marketing with an already approved innovator drug product to evaluate the safety/efficacy of the new formulation drug product. E. To provide information on product quality and performance when there are changes in components, composition, and method of manufacture after approval of the drug product. |
| Measurement | Bioavailability is an indirect or surrogate measure of the rate and extent to which the drug substance or active moiety is absorbed from a drug product and becomes available at its target sites of action. Bioavailability is measured by using three main pharmacokinetic variables: AUC, Cmax, Tmax. | Bioequivalence (BE) is a surrogate measure of in vivo drug product performance and dissolution profile comparisons as a measure of in vitro drug product performance. BE has been established via bioavailability testing. Bioequivalence is measured based on the relative bioavailability of the innovator drug product versus the generic drug product. |
| Nature of Study | Expletory study | Confirmatory or statutory study |
| Interrelation | Not assesse via bioequivalence testing. | Assess via bioavailability testing. |
| Examples | After oral administration of 20 mg of Rabeprazole Sodium Delayed-Release tablet, 52% of this drug is absorbed unchanged systematically as compared to intravenous administration. So, the absolute bioavailability of this drug product is 52%. | Based on the bioequivalence studies under fasting and fed conditions, Idiazole, Rabeprazolnatrium, and Rodesa 20 mg tablets are bioequivalent with Pariet 20 mg. Here, Idiazole, Rabeprazolnatrium, and Rodesa 20 mg tablets are the generic drug product. Pariet is the RLD of Rabeprazole Sodium Delayed-Release tablet in the UK and Australia markets. |
References
| 1. Center for Drug Evaluation and Research (2003). “Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products – General Considerations”. United States Food and Drug Administration. |
| 2. Center for Drug Evaluation and Research (2003). “Guidance for Industry: Bioavailability Studies Submitted in NDAs or INDs — General Considerations”. United States Food and Drug Administration. |
| 3. Center for Drug Evaluation and Research (2003). “Guidance for Industry: Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA — General Considerations”. United States Food and Drug Administration. |
| 4. Birkett D. Generics – equal or not? Australian Prescriber, 2003(26):85-7. |
| 5. WHO Guidance for organizations performing in vivo bioequivalence studies. WHO Technical Report Series No. 996, 2016, Annex 9 |
| 6. United States Pharmacopeia and National Formulary (USP 43–NF 38). United States Pharmacopeial Convention; 2020. Accessed January 01, 2021. |
| 7. Highlights of prescribing information of ACIPHEX® (rabeprazole sodium) Delayed-Release Tablets. Eisai Inc. Retrieved 27 February 2021. |