Quality control tests of tablets or evaluation of tablets is a systematic determination of physical, chemical, mechanical, biological, or microbiological properties of tablets on the basis of in-house (Non-Pharmacopoeial), Pharmacopoeial standards such as BP, USP, Ph. Eur., Ph. Int., JP, IP, ChP, or others guidelines such as ICH etc. To design the perfect tablets and later monitor tablet production quality, quality control tests of tablets or evaluation of tablets’ physical, chemical, and bioavailability properties must essential. Bioavailability vs Bioequivalence
Table of Contents
Types of Tests to Evaluate the Qualities of Tablets
A variety of methods are used for the evaluation of tablets or conducting quality control tests of tablets. All of the quality control tests of tablets or evaluation tests of tablets are classified into three categories:
A. Non-Pharmacopoeial or Non-Official Tests or In-House Tests of Tablet:
- Appearance/ Description
- Thickness and Diameter
- Hardness
- Organoleptic properties
B. Pharmacopoeial or Official Tests of Tablets:
- Identification Tests
- Friability Test
- Disintegration Test
- Weight Variation Test
- Uniformity of Dosage Unit Test
- Dissolution Test
- Assay Test
- Impurities Test
C. Specific Pharmacopoeial Tests of Tablets
- Microbiological Examination of Tablets
- Acid-Neutralizing Capacity
- Quality test of Splitting Tablets with Functional Scoring
- Water content
Non-Pharmacopoeial or Non-Official Tests or In-House Tests
The choice of these tests and their specification depend on the formulator during drug product development and these tests are not restricted or specified in any pharmacopoeia.
Appearance/ Description
The appearance of a tablet is crucial for patient compliance and identification. The control of the appearance of a tablet includes the measurement of several attributes such as a tablet’s shape, surface texture, diameter, thickness, color, absence or presence of an odor, taste, physical flaws and consistency, scoreline, and legibility of any unique identification markings such as embossed or engraved with a logo or letter(s).
For example, A light blue colored, vanilla flavored, round, biconvex film-coated tablet with plain surfaces on both sides. Appearance is a non-pharmacopoeial/ unofficial/ in-house evaluation of tablets or quality control tests of tablets.
Unique Identification Markings
Pharmaceutical companies often use some type of unique markings such as embossed or engraved with a symbol or letters or printing on the tablet for rapid identification. The tablets may score in halves or quadrants to facilitate breaking or to make the smaller dose. Intact and clear unique identification markings on tablets are acceptable.
Thickness of tablets
The thickness of the tablet is the only dimensional variable related to the tablet compression process. Generally, it is measured with a micrometer. The thickness should be within ±5% variation of a standard value and must control for patient acceptance and make the tablet packaging easier.
Diameter and Shape of Tablets
The diameter and shape of the tablets should be controlled by the diameter and shape of the die and punches during the compression process. USFDA recommends that the diameter of the tablet should be 8 mm or less than 8 mm and should not exceed 22 mm. Generally, tablet shapes are round, oval, oblong, caplet, cylindrical, triangular etc. The upper and lower surfaces of tablets may be flat, round, concave, or convex to various degrees. The diameter and shape of the tablet influence esophageal transit, administration techniques (i.e., use of fluids, patient position), and irrespective of patient factors. 21 Core Tablet defects
USFDA recommends that the diameter of the tablet should be 8 mm or less than 8 mm and should not exceed 22 mm.
Organoleptic properties (color, odor, and taste)
Color: Tablet color is crucial for identification and patient acceptance.
Odor: Some types of tablets such as ODT tablets, and chewable tablets have an odor to make a pleasant taste and improve patient acceptance. Besides in some tablets, flavoring agents are used within coating material to mask bad odor.
Taste: Taste is important for patient acceptance, especially for ODT tablets, chewable tablets, and dispersible tablets.
Tablet breaking force/Hardness of Tablets
The breaking force of tablets is commonly called “hardness” in the pharmaceutical literature; however, the use of this term is misleading according to USP. Certainly, tablets require a definite amount of hardness to withstand mechanical shocks of handling in manufacture, packaging, and transportation without affecting the disintegration limit. Generally, oral tablets have a hardness of 4 to 10 kg. However, ODT tablets and chewable tablets have less hardness and often sustained-release tablets are much harder. The units of measurement of tablet hardness are Kilogram (kg), Newton (N), Kilopond (kp), Pound (lb), and Strong-Cobb (SC).
Pharmacopoeial or Official Tests for the Evaluation of tablets
These tests are specified either in individual product monographs or general monographs.
Identification Tests of Tablets
The identification test is specified in a product monograph as an aid to confirm that the tablet contains the labeled drug substance by providing positive identification of the active substance(s) and identification of specific excipient(s), such as preservatives in a drug product.
Generally, one method of confirming the identity is to compare the retention time of the sample with that obtained for the standard injections in a chromatographic assay procedure by HPLC. Other methods also used to orthogonally confirm the identity of the active ingredient are: Thin-Layer Chromatographic Identification Tests, Spectroscopic Identification Tests, Nuclear Magnetic Resonance Spectroscopy, Near-Infrared Spectroscopy as well as Raman Spectroscopy among others [2]. It is a pharmacopoeial test for the evaluation of tablets or quality control tests of tablets.
Friability test of uncoated tablets
Friability testing is used to test the durability of tablets during transit (packing, transportation). Measurement of tablet friability supplements other physical strength measurements, such as tablet breaking force. It is a pharmacopoeial test for the evaluation of tablets or quality control tests of tablets. You may also read the Friability test of tablets, Granules, and Spheroids.
Friability test formula
For ≤ 650 mg weight of tablets, take 6.5 g tablets or as near as possible to 6.5 g. For tablets with more than 650 mg weight, take 10 tablets. The tablets must be carefully dedusted before testing. Friability may be calculated from the following equation:

Friability test limit: A maximum weight loss (obtained from a single test or from the mean of three tests) of not more than 1.0% is considered acceptable [2, 3]. Moreover, effervescent tablets and chewable tablets may have different specifications as far as friability is concerned.
Disintegration Time Test
Disintegration is the process by which a solid oral dosage form such as a tablet breaks down into smaller particles or granules. The tablets must disintegrate and all particles must pass through the 10-mesh screen in the time specified. Complete disintegration is that state in which any residue of the unit (tablet or capsule or granules) except fragments of insoluble coating or capsule shell, remaining on the screen of the disintegration apparatus or adhering to the lower surface of the disk if used, is a soft mass having no palpably firm core. Disintegration is provided to determine whether tablets, capsules, or granules disintegrate within the prescribed time when placed in a suitable liquid medium in a 1000 ml beaker at 37°C ± 2°C. It is a pharmacopoeial test for the evaluation of tablets or quality control tests of tablets.

Disintegration Time of Tablets as per USP
| Types of Tablet | Immersion Fluid/ Medium | Temp. | Limit |
| *Uncoated or Plain-coated tablets | Water or specified medium as the immersion fluid. | 37 ± 2°C | As specified in the individual product monograph. |
| Delayed-Release Tablets or Acid-Resistant or Enteric-Coated Tablet | 0.1 M Hydrochloric acid, or Simulated Gastric Fluid as specified in the monograph. | 37 ± 2°C | After 1 hour no evidence of disintegration, cracking or softening. |
| pH 6.8 phosphate buffer, or Simulated Intestinal Fluid as specified in the product monograph. | 37 ± 2°C | As specified in the individual product monograph. | 5 mins or as specified in the individual product monograph. |
| Effervescent Tablets | 200 ml of water in 250–400 ml beaker. | 37 ± 2°C | 5 mins or as specified in the individual product monograph. |
| Effervescent Granules | Place 1 dose in 200 ml of water in 250–400 ml beaker. | 37 ± 2°C | 5 mins or as specified in the individual product monograph. |
| Buccal, Sublingual Tablets, Orally Disintegrating Tablets, Chewable Tablets | Water or specified medium as the immersion fluid. | 37 ± 2°C | As specified in the individual product monograph. |
| Tablets for Oral Suspension or Oral Solution or Topical solution | Water or specified medium as the immersion fluid. | 37 ± 2°C | As specified in the individual product monograph. |
Disintegration Time of Tablets as per BP
| Types of Tablets | Immersion Fluid/ Medium | Temp. | Limit | Remarks |
| The limit is typically 2 h to 3 h but even with authorized deviations is not less than 1 h. | Water | 37 ± 2°C | 15 Minutes | If fail to comply repeat the test on a further 6 tablets, omitting the discs. |
| Film Coated Tablets | Water | 37 ± 2°C | 30 Minutes | If fail to comply repeat the test on a further 6 tablets, omitting the discs. |
| Coated Tablets other than Film-Coated Tablets / Sugar Coated Tablets | Water | 37 ± 2°C | 60 Minutes | If fail to comply repeat the test on a further 6 tablets, replacing water with 0.1 M hydrochloric acid. If 1 or 2 tablets fail repeat the test on further 12 tablets. |
| Gastro-Resistant Tablets | 0.1 M Hydrochloric Acid | 37 ± 2°C | After 2 h-3h no evidence of disintegration | Limit is typically 2 h to 3 h but even with authorized deviations is not less than 1 h. |
| pH 6.8 Phosphate Buffer | 37 ± 2°C | 60 Minutes | If fail to comply repeat the test on a further 6 tablets, omitting the discs. | Place 1 tablet in a beaker. Repeat the operation on 5 other tablets. |
| Effervescent Tablets | 200 ml of Water | 15-25 °C | 5 mins or as specified in the individual product monograph. | Place 1 tablet in a beaker. Repeat the operation on 5 other tablets. |
| Oral Lyophilisates | 200 ml of Water | 15-25 °C | Within 3 Minutes | Place 1 tablet in a beaker. Repeat the operation on 5 other tablets. |
| Soluble Tablets | Water | 15-25 °C | Within 3 Minutes | Soluble tablets are uncoated or film-coated tablets. |
| Dispersible Tablets | Water | 15-25 °C | Within 3 Minutes | Fineness of dispersion: Place 2 tablets in 100 mL of water and stir until completely dispersed and passes through a sieve mesh 710 µm. |
| Orodispersible Tablets | Water | 15-25 °C | Within 3 Minutes | Orodispersible tablets are uncoated tablets intended to be placed in the mouth where they disperse rapidly before being swallowed. |
Uniformity of Weight (Mass) of Tablet
A weight variation test is performed to determine the consistency of formulated preparations. It is a pharmacopoeial test for the evaluation of tablets or quality control tests of tablets. According to USP, BP & IP the accepted limit of weight variation is given below:
| IP/BP | Average Mass Limit | USP |
| Tablet weight 80 mg or less | ± 10% | Tablet weight 130 mg or less |
| More than 80 mg or Less than 250mg | ± 7.5% | 130 mg to 324 mg |
| 250 mg or more | ± 5% | More than 324 mg |
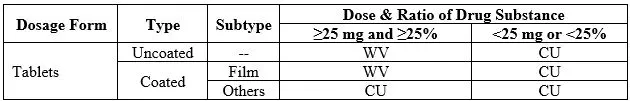
Uniformity of Dosage Unit test for the Evaluation of tablets
The term “uniformity of dosage unit” is defined as the degree of uniformity in the amount of the drug substance among dosage units. To ensure the consistency of dosage units, each unit in a batch should have a drug content within a narrow range around the label claim. It is a pharmacopoeial test for the evaluation of tablets or quality control tests of tablets.
The uniformity of dosage units can be demonstrated by either of two methods, Content Uniformity or Weight Variation. The test for Content Uniformity of preparations presented in dosage units is based on the assay of the individual content of drug substance(s) in a number of dosage units to determine whether the individual content is within the limits set [2,3].
You may apply the content uniformity method in all cases. In addition, which method you select either Content Uniformity (CU) or Weight Variation (WV) to determine the uniformity of dosage unit depends on the dose and ratio of drug substance in a dosage form.
Dissolution test of Tablets
Dissolution is the process in which a substance forms a solution. In vitro, dissolution testing measures the extent and rate of solution formation from a dosage form (the amount of percentage of the drug substance in a dosage form such as tablets, or capsules to go into solution) within a specific time under a specified set of conditions. The terms dissolution and drug release are used interchangeably. The USP dissolution test in the monograph is related to the Bioavailability and Bioequivalence study only when closely allied with a sound regulatory determination. Without this association, the dissolution test should be regarded solely as a quality control test for batch release [4]. It is a crucial pharmacopoeial test for the evaluation of tablets or quality control tests of tablets.
The volume of the dissolution medium is generally 500, 900, or 1000 ml. The use of a hydro-alcoholic medium is discouraged. Certainly, conduct all dissolution tests for IR dosage forms at 37±0.5°C. You may find the dissolution data of drug products at: https://www.accessdata.fda.gov/scripts/cder/dissolution
Before the dissolution test you have to consider the following information:
- Apparatus
- Speed (RPMs)
- Medium Volume (mL)
- Recommended Sampling Times (minutes)
Types of Dissolution Apparatus
The United States pharmacopeia and British pharmacopoeia recommend four types of dissolution apparatus for Test for Solid Dosage Forms:
| General Name | Types of Dissolution Apparatus | Critical test parameters | Dosage form to be tested |
| Apparatus-1 | Basket Apparatus | Rotation Speed | Conventional/Immediate-Release, Prolonged/Extended-Release, Delayed-Release Dosage Forms |
| Apparatus-2 | Paddle Apparatus | Rotation Speed | Conventional/Immediate-Release, Prolonged/Extended-Release, Delayed-Release Dosage Forms |
| Apparatus-3 | Reciprocating Cylinder | Dip Rate | Conventional/Immediate-Release, Prolonged/Extended-Release, |
| Apparatus-4 | Flow-Through Cell | Flow Rate Of Medium | Conventional/Immediate-Release, Prolonged/Extended-Release, |
The acceptable limit of Dissolution
Generally, Not less than 75% (Q) of the labeled amount of the drug is dissolved in 45 minutes or specified in individual product monographs.
For rapidly dissolving products, the generation of an adequate profile sampling at 5- or 10-minute intervals may be necessary. For highly soluble and rapidly dissolving drug products (BCS classes 1 and 3), a single-point dissolution test specification of NLT 85% (Q=80%) in 60 minutes or less is sufficient as a routine quality control test for batch-to-batch uniformity. On the other hand, for slowly dissolving or poorly water-soluble drugs (BCS class 2), a two-point dissolution specification, one at 15 minutes to include a dissolution range (a dissolution window) and the other at a later point (30, 45, or 60 minutes) to ensure 85% dissolution, is recommended to characterize the quality of the product [4].
Dissolution testing and interpretation can be continued through three stages if necessary. In stage 1(S1), six tablets are tested and are acceptable if all of the tablets are not less than the monograph tolerance limit (Q) plus 5%. If the tablets fail S1, an additional six tablets are tested (S2). The tablets are acceptable if the average of the twelve tablets is greater than or equal to Q and no unit is less than Q minus 15%. If the tablets still fail the test, an additional 12 tablets are tested. The tablets are acceptable if the average of all 24 tablets is greater than or equal to Q and if not more than 2 tablets are less than Q minus 15% [2].
Assay test: crucial quality control tests of tablets
The assay is a specific and stability-indicating test to determine the potency (content) of the drug product. The assay of tablets expresses in the terms of grams, milligrams, or micrograms of drug per tablet. It is a crucial pharmacopoeial test for the evaluation of tablets or quality control tests of tablets. The assay limit is mentioned in the individual product monographs.
Generally, according to BP, the Assay limit is between 95.0% and 105.0%. For example, Acetaminophen Tablets contain not less than 95.0% and not more than 105.0% of the labeled amount of paracetamol. According to USP, the Assay limit is between 90.0% and 110.0%. For example, Acetaminophen Tablets contain not less than 90.0 % and not more than 110.0 % of the labeled amount of acetaminophen.
Impurities determination
Impurities in tablets are specified in an individual product monograph or may calculate by ICH Q3B(R2) guidelines. During product manufacture and over the shelf life of the product, impurities may come from the degradation of the drug substance or from interactions between the drug substance and excipient(s), among other factors. It is a crucial pharmacopoeial test for the evaluation of tablets or quality control tests of tablets. Besides process impurities, synthetic by-products, and other inorganic and organic impurities may be present in the drug substance and in the excipients used in the manufacture of the drug product.
Specific Pharmacopoeial Tests of Tablets
Microbiological Examination of Tablets (Nonsterile Products)
This test is used to determine the absence or limited occurrence of specified micro-organisms that may be detected under the conditions described. Some liquid oral products can be subject to extreme microbiological control, and others require none. Generally, the microbial content test should not be required for most of the tablets except vitamin tablets, and sugar-containing tablets. It is a crucial pharmacopoeial test for the evaluation of tablets or quality control tests of tablets.
Acceptance Criteria for Microbiological Quality of Nonsterile Dosage Forms [2]
| Route of Administration | Total Aerobic Microbial Count (CFU/g or CFU/mL) | Total Combined Yeasts/Molds Count (CFU/g or CFU/mL | Specified Microorganism(s) |
| Non-aqueous preparations for oral use | 103 | 102 | Absence of Escherichia coli (1 g or 1 mL) |
Acid-Neutralizing Capacity
Acid-Neutralizing Capacity is a pharmacopoeial test for the evaluation of tablets or quality control tests of tablets. Certainly, this test is applicable only to measure the acid-neutralizing capacity of an antacid tablet.
Limit: NLT 5 mEq of acid is consumed by the minimum single dose recommended in the labeling, and the number of mEq calculated by the formula [2]: Result = [0.8 × (FM × M)] + [0.9 × (FC × C)]
Quality test of Splitting Tablets with Functional Scoring
This test indicates that the label claim of the split portions should be a simple fractional part of the claim for the intact tablet based on the number of scores and the size of the split portion (for example, one-half, one-third, or one-quarter).
An acceptable tablet breaks into the designed number of segments, and each split portion has NLT 75% and NMT 125% of the expected weight of the split tablet portion. NLT 28 of the 30 tablets is acceptable [2]. It is a pharmacopoeial test for the evaluation of tablets or quality control tests of tablets only for scored tablets.
Water content determination
The water content of tablets before and after stability study at specified temperatures and humidity for a fixed time may determine to find out moisture impact on tablets. Generally, water content calculated by using the method is Karl Fischer titration.
In summary, by using the above quality tests you may evaluate tablets, and after satisfactory results, you may release a manufacturing batch of tablets. If you have any question comments in the comments section and stay connected with pharmaeducation.net
Keywords: Quality control tests of tablets or Evaluation of tablets, Quality control tests of tablets or Evaluation of tablets, Quality control tests of tablets, Evaluation of tablets or Quality control tests of tablets, Quality control tests of tablets or Evaluation of tablets.
References
1. Center for Drug Evaluation and Research (CDER) (2015). “Guidance for Industry: Size, Shape, and Other Physical Attributes of Generic Tablets and Capsules”. United States Food and Drug Administration.
2. United States Pharmacopeia and National Formulary (USP 43–NF 38). United States Pharmacopeial Convention; 2020.
3. British Pharmacopoeia Commission. British Pharmacopoeia 2021. London: TSO; 2021.
4. Center for Drug Evaluation and Research (CDER) (1997). Guidance document: Dissolution Testing of Immediate Release Solid Oral Dosage Forms”. United States Food and Drug Administration.